
Со едукативната кампања „Ги запознаваме ретките болести“ на Здружението за лица со ретки болести „Живот со предизвици“, секој ден во февруари се зборува за една ретка болест, начинот на дијагностицирање, третман и предизвиците со кои се соочуваат луѓето што живеат со неа. Како ја добиваат дијагнозата? Дали има некаков лек? Колку и дали воошто се пристапни и достапни лековите? – се прашањата кои се откриваат во кампањата.
Merosin-deficient congenital muscular dystrophy
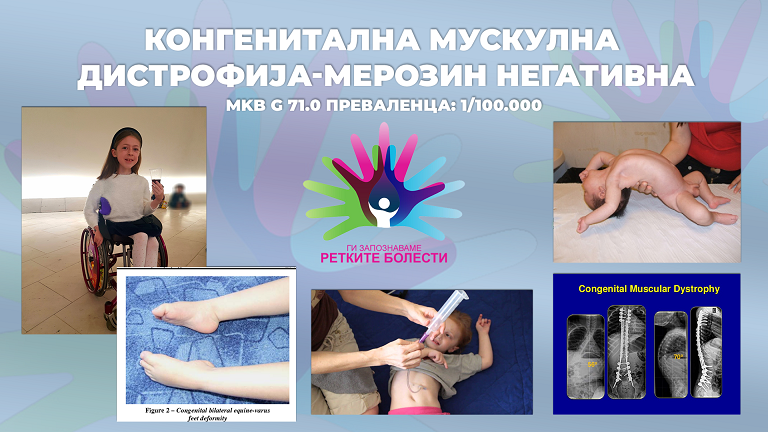
Конгенитална мускулна дистрофија-Мерозин негативна
МКБ Г 71.0
Преваленца (распространетост) : 1/100.000
Конгенитална (вродена) мускулна дистрофија (ЦМД1А) претставува генетско хетерогено невромускулно нарушување кое се јавува при самото раѓање или веднаш по него и ги зафаќа мускулите, па ги ослабува и како резултат на тоа со текот на времето, тие почнуваат да се губат. Мерозин-негативна конгенитална мускулна дистрофија или ЦМД1А, е една од најчестите видови на конгениталната мускулна дистрофија. Предизвикана од ретка генетска мутација на генот Лама-2, одговорен за протеинот мерозин околу мускулното влакно, претставува тешка форма на дистрофија карактеризирана со тешка мускулна хипотонија-намален мускулен тонус при раѓање или веднаш по раѓањето, прогресивна слабост на мускулите и дегенерација-атрофија, одложени моторни функциии со неможност за стоење и одење, прогресивни контрактури на зглобовите со варијабилна сериозност, нормална интелегенција, иако во некои случаи се појавува и умерена ментална ретардација и епилепсија.
Застапеност на болеста:
ЦМД1А ја зафаќа женската и машката популација подеднакво. Точната застапеност не е позната поради недостаток на дијагностичка генетска потврда во минатото, но повеќе истражувања укажуваат дека едно на 100.000 бебиња е заболено со оваа болест. Во нашата држава, од досегашните сознанија, само едно бебе е родено со оваа болест до сега.
Историја и терминологија:
Конгениталната мускулна дистрофија за прв пат била опишана во 1903 година од Фредерик Еустас Батен како „инфантилна миопатија“, па со подетални хистолошки набљудувања во доцните 60-ти години на ЏЏ век, се заменува со сегашниот термин „вродена“. Но, напредокот бил релативно бавен сè до 1990 година. Со големи достигнувања на биохемиска и молекуларна основа, во 1995 година се идентификувани првите генетски мутации што доведуваат до откривање на целосен недостаток на протеинот мерозин. Врз основа на генетски откривања, Конгенитална мускулна дистрофија е поделена на подвидови според генетскиот и протеинскиот недостаток, па така и мерозин-негативната конгенитална мускулна дистрофија (ЦМД1А) е именувана по генетскиот недостаток на протеинот мерозин.
Причина за појава на болеста:
ЦМД1А се наследува на автосомно-рецесивен начин, а тоа значи дека мора да има две патогени мутации кои предизвикуваат појава на симптоми. Кај оваа ретка болест има две мутации во генот ЛАМА2 (ламинин алпха 2, лоциран на 6 љ 22-23 и има 64 егзони) кој го произведува протеинот мерозин, еден од протеините на ламининот кој е составен дел од скелетните мускули и периферните нерви и претставува дел од мускулната клеточна мембрана и кој влијае врз стабилноста на мускулната клетка и мускулните влакна и помага во врзување на другите протеини во мускулната клеточна мембрана.Овие мутации се наследуваат од двајцата родители или може да бидат спонтано настанати. За едно дете да биде засегнато со ЦМД1А, и двајцата родители мораат да бидат носители на истата мутација на генот ЛАМА-2 со што и двајцата ќе ја пренесат мутацијата на генот на своето дете. Детето наследува два изменети ЛАМА-2 гена, еден од мајката и еден од таткото. Секој од родителите на поединецот со ЦМД1А, има по една копија на променетиот ген ЛАМА-2 и е познат како носител, но кај носителот не се пројавуваат знаци и симптоми од болеста. Родителите-носители имаат 25-процентна шанса да добијат дете заболено од ЦМД1А, 2 од 4 шанси да добијат дете само како носител на мутираниот ген т.е. 50 % и 1 од 4 шанси т.е. 25 % да добијат дете кое нема да го носи мутираниот ген.
Клиничка слика:
Бебињата со ЦМД1А наречени „флоппѕ бабѕ“, се препознатливи уште по самото раѓање по нивната позиција на лежење. Тие не можат да се мрдаат како другите бебиња поради тешката хипотонија-слабиот мускулен тонус, плачот им е многу слаб, немаат стисок на рацете, не можат да ја држат главата, имаат високо покачена серумска-креатин кинеза (ЦК), респираторна инсуфициенција и потреба од респираторна поддршка. Пациентите со оваа ретка болест имаат прогресивно-слаба мускулатура, мускули кои со текот на времето ќе бидат послаби и ќе почнат да се губат, одложени моторни функции, неможност за седнување/станување и одење со што стануваат зависни од инвалидски колички, имаат вродени или стекнати контрактури кои ќе прогресираат поради задебелување и скратување на ткивото и ќе предизвикаат поголеми деформитети и ограничувања во движењата поради што станува нужно користењето на ортопетски помагала и лонгети, вертикализатор, секојдневно вежбање со физиотерапевт и хидротерапија. Ослабувањето на мускулите ќе доведе до сериозно влошување на цврстината на ‘рбетниот столб и појава на прогресивна сколиоза за којашто ќе биде неопходна операција. Телесната тежина кај овие пациенти е доста мала. Со текот на времето ослабуваат и респираторните мускули, па како резултат ќе се појават пулмолошки инфекции, намален кислород и потешкотии во ноќното дишење за што ќе биде потребно приклучување на респираторни машини и вентилатор. Прогресивни проблеми се јавуваат и при хранењето за што е потребно вметнување на цевка за гастростомија. И покрај присутната промена на белата маса во мозокот, кај овие пациенти менталната интелегенција е нормална, но има и мал број на пациенти каде се забележани потешкотии при учење и епилептични напади.
Дијагностика:
Дијагнозата на ЦМД1А бара истовремена експертиза во повеќе специјалности (неврологија, морфологија, генетика, неврорадиологија), достапна во неколку центри ширум светот кои имаат доволно искуство со различните подвидови на КМД. По утврдување на хипотонија, докторите најпрво прават специјални тестови за нервна спроводливост и електромиографија (ЕМГ) кои се користат за стимулирање и процена на мускулите или нервите индивидуално за да се согледа проблемот за хипотонијата. Во почетната фаза на дијагностичкиот процес, лекарите често препорачуваат посебен тест на крвта- Серумска Креатин Кинеза(ЦК) кој се однесува на ензим што протекува надвор од оштетениот мускул. Покаченото ниво на ЦК значи дека мускулите се уништуваат со некој абнормален процес, како што е мускулна дистрофија или воспаление. По ова следува мускулна биопсија при која од земено парче мускул се прават комплексни генетски испитувања со што ќе се утврди точната дистрофија и кодот на мутацијата на ЛАМА-2 генот. Со добиениот код, семејството ќе има можност да направи понатамошни пренатални испитувања за следните бремености и да утврди дали плодот ја има истата мутација на генот и ризикот за заболување од ЦМД1А.
Третман:
Сè уште нема пронајден лек или третман за оваа ретка болест. Во 2008 год. Омигапил од Сантхера доби одобрување како лек за ЦМД1А и ја помина првата фаза со што продолжи испитувањето. Сè уште се работи на третманот Ламинин-111 кој до сега покажа успорување на прогресивната мускулна слабост на моделот-глушец. Од обезбедувањето на сите неопходни машини, помагала и квалитетна физио-хидро терапија, ќе зависи квалитетот и должината на животот на овие пациенти за кои важи дека имаат скратен животен век.
За проектот:
Гордана Лолеска, Супер радио и „Живот со предизвици“ и овој февруари Ве запознаваат со ретките болести во Република Македонија со надеж дека преку овие 28 ретки дијагнози сите заедно ќе научиме нешто повеќе.
Првиот ден на ретки болести беше одбележан во 2008 година, точно на 29 февруари, „редок“ датум кој се случува само еднаш на секои четири години. Од тогаш, Денот на ретки болести се одржува секоја година на последниот ден од февруари, месец познат по тоа што има „редок“ број на денови.
Денот на ретките болести е меѓународна кампања за подигнување на свеста за ретките болести. Од почетокот во 2008 година, илјадници настани се организираат низ целиот свет и опфаќаат милиони луѓе. Кампањата започна како европски настан и со текот на времето стана глобален феномен, со учество на над 90 земји од целиот свет во 2018 година.
Република Македонија за прв пат се вклучи во одбележување на овиј редок ден во 2013 година.
Кампањата за Денот на ретки болести е за пошироката јавност, за релевантните институции и секој е добредојден да се придружи: пациенти и семејства, организации на пациенти, медицински професионалци, истражувачи, фармацевтска индустрија, јавни здравствени институции – колку повеќе не има, толку подобро. Денот на ретките болести е можност за учесниците да бидат дел од глобалниот повик кон креаторите на политики, истражувачите, компаниите и здравствените професионалци повеќе и поефикасно да ги вклучат пациентите во истражувањата за ретки болести.
Republika.mk - содржините, графичките и техничките решенија се заштитени со издавачки и авторски права (copyright). Крадењето на авторски текстови е казниво со закон. Дозволено е делумно превземање на авторски содржини (текст и фотографии) со ставање хиперлинк до содржината што се цитира.

Comments are closed for this post.